Design and synthesis of highly potent third generation inhibitors of Arginase I and II
L-arginine metabolism is emerging as a crucial regulatory pathway for a wide variety of oncological,1 autoimmune,2 anti-inflammatory3 and infectious diseases.4 Arginase, which competes with inducible nitric oxide synthase (iNOS) for L-arginine as a common substrate, is dysregulated in many such diseases. The diverse pathologies associated with increased arginase expression levels arise from both increased production of ornithine, proline and polyamines, the products of arginase-mediated hydrolysis of arginine, and the decreased nitric oxide (NO) production that results from reduced substrate availability for iNOS.
Scheme 1.

Evaluation of ABH and our related second-generation seriesof analogs5 co-crystalized with arginase indicated a five-memberedring connecting the amino acid carbon with the first carbon on the butane boronic acid side chain could potentially provide a suitable constraint without introducing negative Van der walls interactions with active site amino acids, provided the amine and boronic acid side chain were positioned in an anti-orientation as illustrated in Scheme 1. In addition to reducing entropy, the atoms used to create the ring constraint could be used as a scaffold to introduce additional substituents that would form hydrogen bonding interactions with Asp 180 and Asp 182 in a manner analogous to our previous series5. A pyrrolidine-based ring system was selected because it would provide convenient access to a versatile late-stage intermediate that would accelerate the lead optimization process.
Synthesis of these pyrrolidine-based arginase inhibitors was completed using the chemistry outlined in Scheme 2. Addition of allyl magnesium bromide to commercially available boc-protected pyrrolidine epoxide 1 followed by oxidation with sulfur trioxide pyridine complex gives allyl ketone 2 in 79% yield (2 steps). The racemic amino acid moiety 3 is formed via the Ugi reaction using our standard conditions. The desired anti-diastereomer is separated into its enantiomers using a classical resolution with dibenzoyl-L-tartrate [(2S,3S)-2,3-bis(benzoyloxy)-4-(isopropylamino)-4-oxobutanoic acid]. Using this method, the desired enantiomer 5 is obtained in approximately 77% theoretical yield with an enantiomeric excess of 99.7% as determined by chiral HPLC after reintroduction of the boc-group. Subsequent hydroboration and hydrolysis results in the unsubstituted pyrrolidine 7 in 68%% yield (2 step).
Scheme 2.
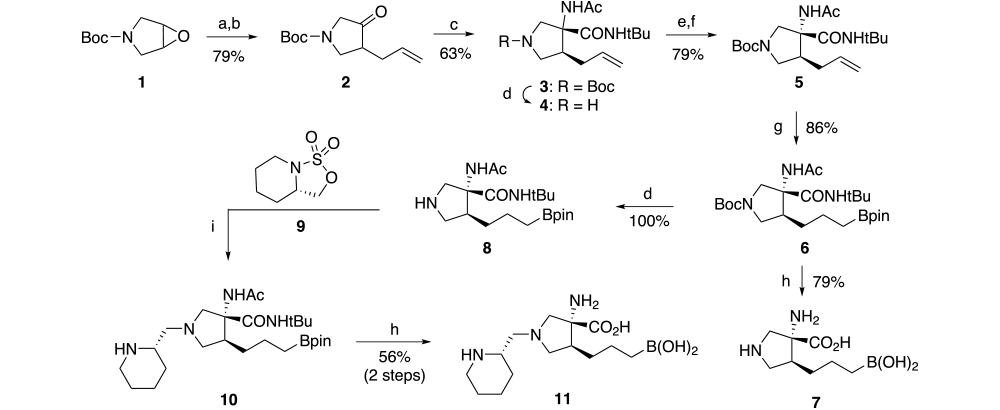
Reagents and conditions: (a) allyl magnesium bromide, ether, 0 oC; (b) pyridine-sulfurtrioxide, DMSO, diisopropylethylamine, DCM, 0 oC; (c) t-BuNC, NH4Ac, CF3CH2OH, RT, 3 days; (d) TFA, DCM; (e) (2S,3S)-2,3-bis(benzoyloxy)-4-(isopropylamino)-4-oxobutanoic acid, MeOH, IPA; (f) BOC2O, aq NaHCO3, EtOAC; (g) pinicolborane, Ir2Cl2(COD)2, dppe, THF; (h) 9N HCl, AcOH, 100 oC, 12 h; (i) ACN.
Boc-protected intermediate 6 can be selectively deprotected using trifluoroacetic acid to give the corresponding amine (8) which is a versatile intermediate for use in reductive amination and alkylation reactions. When the desired aldehyde, such as (S)-piperidine-2-carbaldehyde, contains an adjacent chiral center, the reductive amination reaction conditions give primarily the epimerized product. To avoid this racemization, the substituted ethylamine moiety can be introduced using alkylation chemistry via the corresponding cyclic sulfamate (9) which is prepared from (S)-piperidin-2-ylmethanolusing the general method described by Alker6. Alkylation of pyrrolidine intermediate 8 with the desired sulfamate (9) in acetonitrile gives alkylation product 10. Subsequent hydrolysis gives the target amino acid (11) as a single enantiomer in 10 steps with an overall yield of approximately 10%.
Analysis of compound 11 co-crystalized with hArg II confirms the hydrogen bonding interaction between the piperidine ring nitrogen and Aspartic acid residues. As illustrated in Figure 1, the piperidine nitrogen forms one hydrogen bond with Asp 200 (3.09 A) and one with Asp 202 through a water molecule (2.73 A).
Consistent with the X-ray crystalography data, screening results for compound 11 indicate strong binding interactions between the inhibior and active site amino acids. Arg I and Arg II IC50s for compound 11 are 2.6 and 14 nM respectively (Scheme 3).
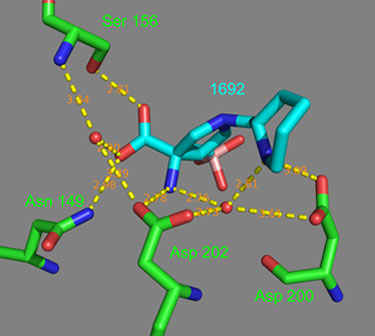
Figure 1.
Example 11 (blue) bound in the arginase II active site pocket (green) with ionic and hydrogen bonds (yellow) and bond distances (salmon).
Scheme 3.
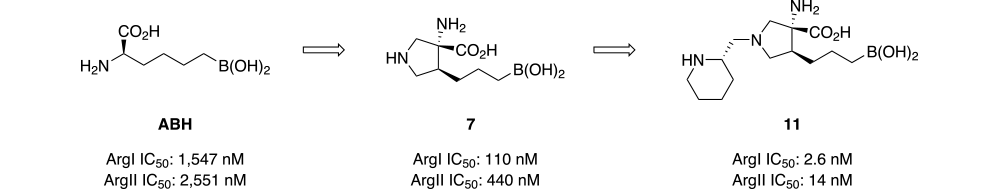
The increased potency relative to ABH arises from the added effects of two specific structural changes: introduction of a ring constraint that reduces entropy, and addition of a basic amine side chain that is positioned to form new ionic interactions with aspartic acid residues in the active site pocket. This new class of arginase inhibitors is significantly more potent than any series previously reported. It is hoped that these compounds will help facilitate a better understanding of arginase and the specific pharmacological role it plays so many important diseases.
References
- (a) Andersen, M. H. The balance players of the adaptive immune system. Cancer Research 2018, 78, 1379-1382., (b) Sawa-Wejksza, K.; Kandefer-Szerszen, M. Tumor-associated macrophages as a target for antitumor therapy. Arcivum Immunologiae et Therapisae Experimentalis 2018, 66, 97-111.
- Pernow, J.; Jung, C. Arginase as a potential target in the treatment of cardiovascular disease: reversal of arginine steal? Cardiovascular Research 2013, 98, 334-343 (b) Ming, X-F.; Yang, Z.. Functions and mechanisms of arginase in age-associated cardiovascular diseases. Current Translational Geriatrics and Experimental Gerontology Reports 2013, 2, 268-274. (c) Tratsiakovich, Y.; Yang, J.; Gonon, A.; T.; Sjoequist, P-O.; Pernow, J. Arginase as a target for treatment of myocardial ischemia-reperfusion injury. European Journal of Pharmacology 2013, 720, 121-123.
- Wegner, A.; Verhagen, J.; Wraith, D. C. Myeloid-derived suppressor cells mediate tolerance induction in antoimmune disease. Immunology 2017, 151, 26-42.
- Antoniu, S.A. Enhancing the nitric oxide synthesis pathway in asthma: a plausible therapeutic approach? Current opinion in investigational drugs 2010, 11, 543-549
- (a) Van Zandt, M. C.; Whitehouse, D.L.; Golebioski, G.; Ji, M.; Zhang, M.; Beckett, P.; Sheeler, R.; Andreoli, M.; Conway, B.; Mahboubi, K.; Mitschler, A.; Cousido-Siah, A.; Xavier, F.; Figueras, R.; Howard, E.; Podjarny, A. D.; Schroeter, H. Discovery of (R)-2-amino-6-borono-2-(2-(piperidin-1-yl)ethyl) hexanoic acid and Congeners as Highly Potent Inhibitors of human Arginase I for Treatment of Myocardial Reperfusion Injury. J. Med. Chem. 2013, 56, 2568-2580 (b) Golebiowski A.; Whitehouse, D.; Beckett, P.; Van Zandt, M.; Ji M.; Ryder, T. R; Jagdmann, E.; Andreoli, M.; Lee Y.; Sheeler, R.; et al. Synthesis of quaternary α-amino acid-based arginase inhibitors via the Ugi reaction Synthesis of quaternary a-amino acid-based arginase inhibitors via the Ugi reaction. Bioorganic & medicinal chemistry letters 2013, 23, 4837-4841.
- Alker, D.; Doyle, K. J.; Harwood, L. M.; McGregor, A. The direct synthesis of the cyclic sulfamidate of (S)-prolinol: simultaneous N-protection and activation towards nucleophilic displacement of oxygen. Tetrahedron Asymmetry 1990, 1, 877-880.